

2020-04-14编辑:极客基因点击:1505
单细胞RNA测序(scRNA-seq)已成为评估免疫细胞异质性的常用工具。近期开发的一些新方法(如CITE-seq和REAP-seq)能够平行测定蛋白质和转录本水平,但这些方法可能需要大量的资源,例如每个细胞所需的测序深度要增加一倍。
为此,弗雷德•哈钦森癌症研究所的研究人员开发出一种新方法,来测定单个细胞内的蛋白质表达和RNA转录水平。这种方法不需要其他方法那么高的测序深度,不过仍然保留了检测低丰度转录本的高灵敏度。
研究人员在《Cell Reports》杂志上发表了这一成果。他们利用该方法来研究免疫细胞异质性,并采用一种名为One-SENSE的质谱流式数据分析工具对蛋白质-转录本数据集进行可视化。
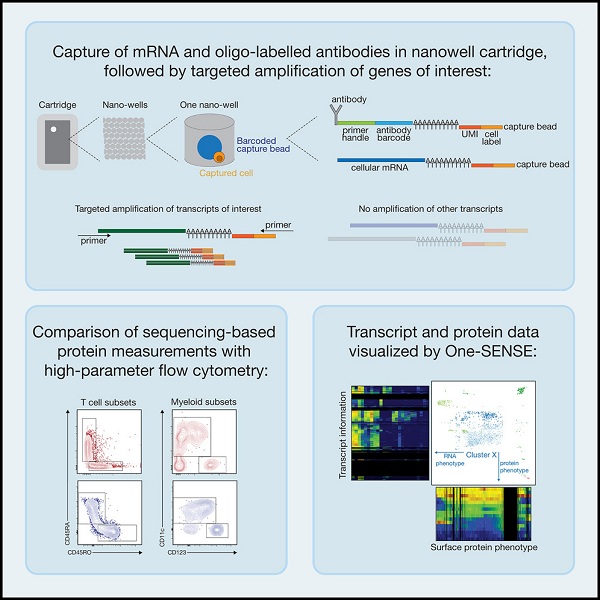
在这项研究中,Florian Mair领导的团队利用带有寡核苷酸条形码的抗体对细胞进行染色,并利用BD Biosciences的Rhapsody平台来捕获纳米孔中的单细胞,以开展靶向转录组学分析。通过这种方式,他们同时拷问了492个免疫相关基因和41种细胞表面蛋白。
为了检验他们的方法,研究人员从三名健康对照身上采集外周血单核细胞样本并分为两批,一批开展多组学分析,另一批利用流式细胞术开展表型分析。他们发现,两种方法都能够在很大程度上区分各个免疫细胞群体。
此外,他们还将这种靶向转录组学方法与常用的全转录组分析方法进行比较,发现这两种方法都可以捕获主要的外周血单核细胞谱系。
研究人员之后又利用一名供体的外周血单核细胞样本(每个细胞约有27,000条序列),看看利用这种多组学方法来解析不同信号所需的读取深度。他们对该数据集中的序列进行二次抽样,发现在使用100%的序列和使用20%的序列时,蛋白质信号之间几乎没有差异。不过,仅使用10%的序列时,信号差异变得很明显。
这些结果表明,对于这种靶向方法,每个细胞需要2,000至4,000条序列,仅仅是全转录组测序方法所需读取深度的十分之一。此外,对于文库中的抗体部分,每个细胞需要200-400条序列/抗体,才能提供足够的分辨率。研究人员估计,他们的方法大约要便宜五倍。
通过修改质谱流式分析工具One-SENSE,研究人员能够对多组学数据集中的转录本和蛋白质表达之间的相关性进行可视化。他们在一个轴上绘制蛋白质表达图谱,在另一个轴上绘制差异表达的基因表达谱。这样很容易识别转录本相似而蛋白不同的细胞簇,反之亦然。将这种方法应用于外周骨髓细胞数据时,他们可以识别CD14+骨髓群体中的不同亚群。
作者表示,他们希望这些结果能够促使更多的科学家采用多组学方法,并在未来推动个性化医学研究。(生物通 薄荷)
原文检索
A Targeted Multi-omic Analysis Approach Measures Protein Expression and Low-Abundance Transcripts on the Single-Cell Level
DOI:https://doi.org/10.1016/j.celrep.2020.03.063
来源:生物通 侵删
关注极客基因公众号
看单细胞测序前沿资讯

